Workflow
One typical approach to detect splicing variants involves utilizing both genomic and transcriptomic data, followed by conducting statistical analyses to correlate genomic variations with changes in the transcriptome (Jung et al., Nature Genetics, 2015; Shiraishi et al., Genome Research, 2018; PCAWG Transcriptome Core Group et al., Nature, 2020). However, this approach requires access to both genome and transcriptome sequencing data, which is often only available in well-coordinated projects like TCGA. Contrastingly, the Sequence Read Archive hosts an extensive collection of publicly accessible transcriptome sequence data, numbering in the hundreds of thousands.

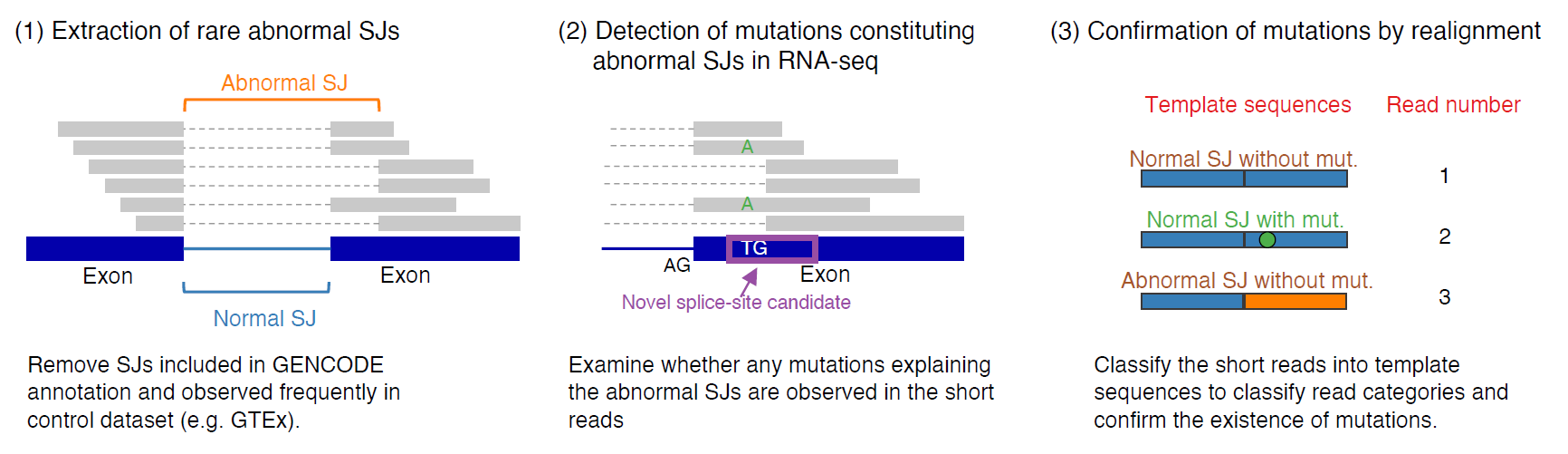
The key observation behind the development of juncmut was "the frequent observation of mismatch bases, indicative of SSCVs, in the short reads of transcriptome sequence data" (Figure 1). This might initially seem contradictory, as one would expect SSCVs, particularly those on the intronic side of a new splice-site, to be spliced out and hence not present in the short reads. Nevertheless, due to the variability in splicing effects and the incomplete dominance of SSCVs (attributable to competition with pre-existing splice-sites), it appears that transcriptome data often exhibits variants supportive of SSCVs in many instances. The overview of juncmut is depicted in the Figure 2.
Main Contributer
Ai Okada
Yuichi Shiraishi
Citation
Please site the following paper when you use the resource in this site.
Iida et al., Systematically developing a registry of splice-site creating variants utilizing massive publicly available transcriptome sequence data, Nat Commun., 2025 Jan 9;16(1):426
.
Notes
Although we conducted screenings of SSCVs using both TCGA and SRA transcriptome data, we chose to exclude those from TCGA. The current algorithm is unable to distinguish between germline and somatic variants, and there is a concern that it might inadvertently lead to the identification of individuals.
Database version and date
ClinVar: 2024-12-23
Cancer Gene Census: 2022-11-29
ACMG SF Gene: v3.2
ClinGen Dosage Sensitivity: 07 Jan, 2025